




Other awesome people in kidney disease research by using deep-learning methods

Vijayakumar Kakade, Yale University
Research Scientist, Internal Medicine (Nephrology). Their group has successfully developed a machine learning algorithm to identify resident kidney cell populations, infiltrating cell populations, and cell activation and injury states in human kidney tissue

Michelle Estrella, University of California, San Francisco
Professor, Medicine. The overall goals of her research are to improve our understanding of chronic kidney disease and its consequences, and to develop strategies to mitigate the risk of chronic kidney disease development and progression

Girish N Nadkarni, Mount Sinai
MD, Nephrology, Internal Medicine.
He is the System Chief of the Division of Data Driven and Digital Medicine (D3M), the Co-Director of the Mount Sinai Clinical Intelligence Center (MSCIC) and the Director of Charles Bronfman Institute for Personalized Medicine.

Amitava Banerjee, University College London
Professor, Clinical Data Science Institute of Health Informatics. His research has investigated quality improvement in CVD, EHR phenotypes, health inequalities and machine learning for subphenotyping and risk prediction

Matthias Kretzler, University of Michigan
Professor of Computational Medicine & Bioinformatics, Internal Medicine. Focus on: Systems biology of renal diseases, including nephrotic syndrome, diabetes, hypertension and autoimmune diseases of the kidney. Coordination of NIH ORDR rare disease Nephrotic Syndrome Study Network for multi scalar analysis of glomerular disease

Julio Saez-Rodriguez, Heidelberg University
Professor of Medical Bioinformatics and Data Analysis at Heidelberg University and Director of the Institute of Computational Biomedicine, a Group Leader at the EMBL- Heidelberg University Molecular Medicine Partnership Unit (MMPU). his computational methods are focusing on his focus is on cancer, autoimmune diseases, and conditions of the kidney and heart

Syed Abdul Shabbir, Taipei Medical University
Professor, Graduate Institute of Biomedical Informatics, Professor Shabbir Syed-Abdul is a strong health informatics research professional with expertise in artificial intelligence and internet of things in healthcare. His research focus revolves around long term care of older adults and early prediction and management of chronic diseases such as cancer, chronic kidney disease, etc

Nam K. Tran, UC Davis Medical Center
Director, Pathology Biorepository; Professor, Division of Clinical Pathology. Dr. Tran has interest in the use of artificial intelligence and machine learning. Specifically, he and his colleagues have pioneered the the development of novel machine learning algorithms for acute kidney injury and sepsis in high-risk burn patients

Azra Bihorac, The University Of Florida
Professor Of Medicine, Surgery And Anesthesiology; Director, Intelligent Clinical Care Center. Her vision is to develop tools for intelligent human-centered health care that delivers optimized care tailored to a patient’s “personal clinical profile” using digital data and work in national and international professional organizations in nephrology and critical care medicine

Joe Ledsam, Google
Research Scientist, Joe is clinician scientist in Google Japan focusing on the application of AI to health and science. Prior to joining Google Japan, Joe spent four years in DeepMind leading multiple research projects across medical imaging and electronic health records, as well as founding the DeepMind Genomics team.

Tomas Ward, Dublin City University
Professor, AIB Chair in Data Analytics
His present research concerns the application of machine learning to improve human health, performance and decision making and am interested in personal analytics approaches

Ayman El-Baz, University of Louisville
Chair, Department of Bioengineering. He has developed a new non-invasive CAD system for the automatic detection of acute renal rejection after kidney transplantation, a noninvasive CAD system that characterizes the status of transplanted kidney based on fusion of stochastic approaches

Spatially resolved transcriptomics provides a new way to define spatial contexts and understand the pathogenesis of complex human diseases. Although some computational frameworks can characterize spatial context via various clustering methods, the detailed spatial architectures and functional zonation often cannot be revealed and localized due to the limited capacities of associating spatial information. We present RESEPT, a deep-learning framework for characterizing and visualizing tissue architecture from spatially resolved transcriptomics. Given inputs such as gene expression or RNA velocity, RESEPT learns a three-dimensional embedding with a spatial retained graph neural network from spatial transcriptomics. The embedding is then visualized by mapping into color channels in an RGB image and segmented with a supervised convolutional neural network model. Based on a benchmark of 10x Genomics Visium spatial transcriptomics datasets on the human and mouse cortex, RESEPT infers and visualizes the tissue architecture accurately. It is noteworthy that, for the in-house AD samples, RESEPT can localize cortex layers and cell types based on pre-defined region- or cell-type-enriched genes and furthermore provide critical insights into the identification of amyloid-beta plaques in Alzheimer's disease. Interestingly, in a glioblastoma sample analysis, RESEPT distinguishes tumor-enriched, non-tumor, and regions of neuropil with infiltrating tumor cells in support of clinical and prognostic cancer applications
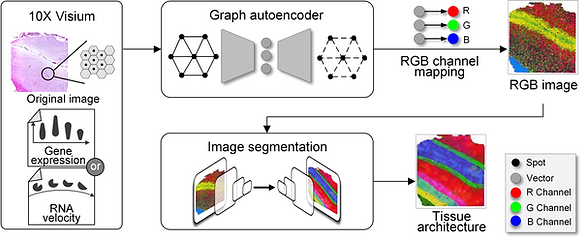
Identifying spatially variable genes (SVGs) is critical in linking molecular cell functions with tissue phenotypes. Spatially resolved transcriptomics captures cellular-level gene expression with corresponding spatial coordinates in two or three dimensions and can be used to infer SVGs effectively. However, current computational methods may not achieve reliable results and often cannot handle three-dimensional spatial transcriptomic data. Here we introduce BSP (big-small patch), a spatial granularity-guided and non-parametric model to identify SVGs from two or three- dimensional spatial transcriptomics data in a fast and robust manner. This new method has been extensively tested in simulations, demonstrating superior accuracy, robustness, and high efficiency. BSP is further validated by substantiated biological discoveries in cancer, neural science, rheumatoid arthritis, and kidney studies with various types of spatial transcriptomics technologies.


High throughput spatial transcriptomics (HST) is a rapidly emerging class of experimental technologies that allow for profiling gene expression in tissue samples at or near single-cell resolution while retaining the spatial location of each sequencing unit within the tissue sample. Through analyzing HST data, we seek to identify sub-populations of cells within a tissue sample that may inform biological phenomena. Existing computational methods either ignore the spatial heterogeneity in gene expression proles, fail to account for important statistical features such as skewness, or are heuristic-based network clustering methods that lack the inferential benefits of statistical modeling. To address this gap, we develop SPRUCE: a Bayesian spatial multivariate finite mixture model based on multivariate skew-normal distributions, which is capable of identifying distinct cellular sub-populations in HST data. We further implement a novel combination of Polya-Gamma data augmentation and spatial random effects to infer spatially correlated mixture component membership probabilities without relying on approximate inference techniques. An R package spruce for fiting the proposed models is available through The Comprehensive R Archive Network (CRAN).


High throughput spatial transcriptomics (HST) technologies have allowed for identification of distinct cell sub-populations in tissue samples, i.e., tissue architecture identification. However, existing methods do not allow for simultaneous analysis of multiple HST samples. Moreover, standard tissue architecture identification approaches do not provide uncertainty measures. Finally, no existing frameworks have integrated deep learning with Bayesian statistical models for HST data analyses. To address these gaps, we developed MAPLE: a hybrid deep learning and Bayesian modeling framework for detection of spatially informed cell spot sub-populations, uncertainty quantification, and inference of group effects in multi-sample HST experiments. MAPLE includes an embedded regression model to explain cell sub-population abundance in terms of available covariates such as treatment group, disease status, or tissue region. We demonstrate the capability of MAPLE to achieve accurate, comprehensive, and interpretable tissue architecture inference through four case studies that spanned a variety of organs in both humans and animal models.


The advent of high throughput spatial transcriptomics (HST) has allowed for unprecedented characterization of spatially distinct cell communities within a tissue sample. While a wide range of computational tools exist for detecting cell communities in HST data, none allow for characterization of community structure – an analysis task that can help elucidate cellular dynamics in important settings such as the tumor microenvironment. To address this gap, we introduce the concept of community connectivity analysis (CCA), which is concerned not only with labeling distinct cell communities within a tissue sample, but understanding the relative similarity of cells within and between communities. We develop a Bayesian multi-layer network model called BANYAN for integration of spatial and gene expression information to achieve CCA. We use BANYAN to implement CCA in invasive ductal carcinoma, and uncover distinct community structure relevant to the interaction of cell types within the tumor microenvironment. Next, we show how CCA can help clarify ambiguous annotations in a human white adipose tissue sample. Finally, we demonstrate improved community detection with BANYAN as a result of considering spatial information via a simulation study based on real sagittal mouse brain HST data.

Kidney injury
Kidney injury, specifically acute kidney injury (AKI), is a significant concern in both medical science and computational biology due to its sudden onset, potential severity, and complex nature. In medical science, it's recognized as a critical condition that can lead to various complications, including chronic kidney disease (CKD) and increased mortality rates.


Deep learning model design
The application of the deep learning model, which is a field of Artificial intelligence (AI), in the context of kidney injury, particularly Acute Kidney Injury (AKI), represents a significant advancement in biomedical informatics. Advanced deep learning model offers innovative solutions for early detection, diagnosis, management, and prognosis of AKI
System-biology analysis
Combining systems biology with deep learning to study GRNs in kidney injury represents a cutting-edge approach to medical research. It holds the promise of unlocking new insights into the disease, leading to better diagnostics, therapeutics, and understanding of kidney pathology. We are addressing challenges in this analysis related to data, model interpretability, and biological evaluation

Dataset
An atlas of healthy and injured cell states and niches in the human kidney
Lake, B.B., Menon, R., Winfree, S. et al. Nature 619, 585–594 (2023). https://doi.org/10.1038/s41586-023-05769-3